Microbiome data analysis
Introduction

MetaLonDA: a flexible R package for identifying time intervals of differentially abundant features in metagenomic longitudinal studies
Ahmed A. Metwally, Jie Yang, Christian Ascoli, Yang Dai, Patricia W. Finn and David L. Perkins.
Microbiome (2018) 6:32. PMID: 29439731 PMCID: PMC5812052 DOI: 10.1186/s40168-018-0402-y
PubMed
Link to MetaLonDA
Abstract
BACKGROUND:
Microbial longitudinal studies are powerful experimental designs utilized to classify diseases, determine prognosis, and analyze microbial systems dynamics. In longitudinal studies, only identifying differential features between two phenotypes does not provide sufficient information to determine whether a change in the relative abundance is short-term or continuous. Furthermore, sample collection in longitudinal studies suffers from all forms of variability such as a different number of subjects per phenotypic group, a different number of samples per subject, and samples not collected at consistent time points. These inconsistencies are common in studies that collect samples from human subjects.
RESULTS:
We present MetaLonDA, an R package that is capable of identifying significant time intervals of differentially abundant microbial features. MetaLonDA is flexible such that it can perform differential abundance tests despite inconsistencies associated with sample collection. Extensive experiments on simulated datasets quantitatively demonstrate the effectiveness of MetaLonDA with significant improvement over alternative methods. We applied MetaLonDA to the DIABIMMUNE cohort ( https://pubs.broadinstitute.org/diabimmune ) substantiating significant early lifetime intervals of exposure to Bacteroides and Bifidobacterium in Finnish and Russian infants. Additionally, we established significant time intervals during which novel differentially relative abundant microbial genera may contribute to aberrant immunogenicity and development of autoimmune disease.
CONCLUSION:
MetaLonDA is computationally efficient and can be run on desktop machines. The identified differentially abundant features and their time intervals have the potential to distinguish microbial biomarkers that may be used for microbial reconstitution through bacteriotherapy, probiotics, or antibiotics. Moreover, MetaLonDA can be applied to any longitudinal count data such as metagenomic sequencing, 16S rRNA gene sequencing, or RNAseq. MetaLonDA is publicly available on CRAN ( https://CRAN.R-project.org/package=MetaLonDA ).
KEYWORDS:
Differential abundance; Longitudinal studies; Metagenomics; Microbiome; Negative binomial distribution; Smoothing splines; Time series
Detection of Differential Abundance Intervals in Longitudinal Metagenomic Data Using Negative Binomial Smoothing Spline ANOVA
Metwally AA, Finn PW, Dai Y, Perkins DL
Proceedings of the 8th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics. PP. 295-304.
ACM Digital Library
Abstract
Metagenomic longitudinal studies have become a widely-used study design to investigate the dynamics of the microbial ecological systems and their temporal effects. One of the important questions to be addressed in longitudinal studies is the identification of time intervals when microbial features show changes in their abundance of functions. We propose a statistical method that is based on a semi-parametric Smoothing Spline ANOVA and negative binomial distribution to model the time-course of the features between two phenotypes. We demonstrate the superior performance of our proposed method compared to the two currently existing methods using simulated data. We present the analysis results of our proposed method in an analysis of a longitudinal dataset that investigates the association between the development of type 1 diabetes in infants and the gut microbiome. The identified significant species and their specific time intervals reveal new information that can be used in improving intervention or treatment plans.
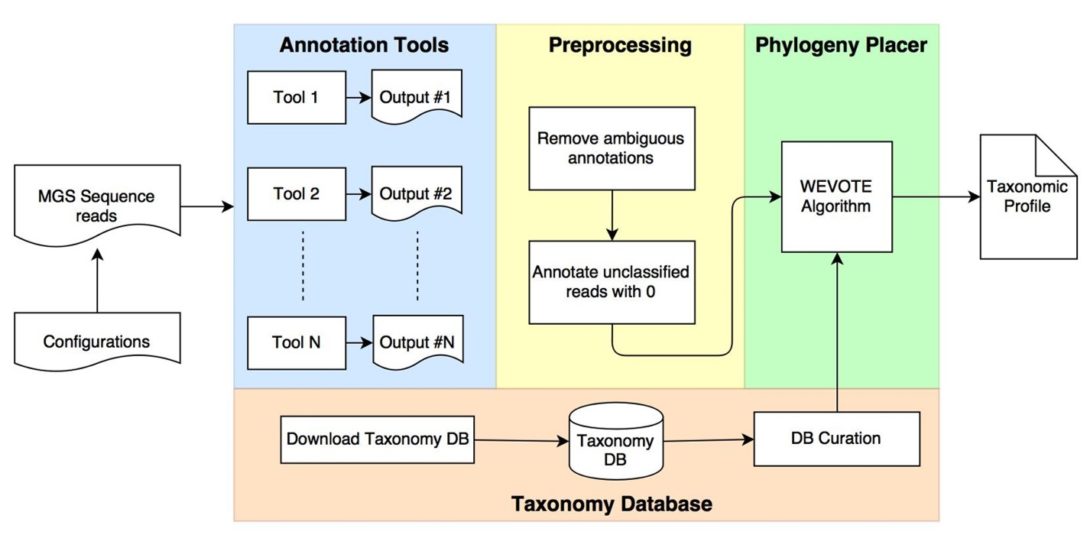
WEVOTE: Weighted Voting Taxonomic Identification Method of Microbial Sequences.
Metwally AA, Dai Y, Finn PW, Perkins DL
PLoS ONE (2016) 11(9): e0163527. doi:10.1371/journal.pone.0163527
PubMed
WEVOTE on GitHub
Abstract
Metagenome shotgun sequencing presents opportunities to identify organisms that may prevent or promote disease. The analysis of sample diversity is achieved by taxonomic identification of metagenomic reads followed by generating an abundance profile. Numerous tools have been developed based on different design principles. Tools achieving high precision can lack sensitivity in some applications. Conversely, tools with high sensitivity can suffer from low precision and require long computation time. In this paper, we present WEVOTE (WEighted VOting Taxonomic idEntification), a method that classifies metagenome shotgun sequencing DNA reads based on an ensemble of existing methods using k-mer-based, marker-based, and naive-similarity based approaches. Our evaluation on fourteen benchmarking datasets shows that WEVOTE improves the classification precision by reducing false positive annotations while preserving a high level of sensitivity. WEVOTE is an efficient and automated tool that combines multiple individual taxonomic identification methods to produce more precise and sensitive microbial profiles. WEVOTE is developed primarily to identify reads generated by MetaGenome Shotgun sequencing. It is expandable and has the potential to incorporate additional tools to produce a more accurate taxonomic profile. WEVOTE was implemented using C++ and shell scripting and is available at www.github.com/aametwally/WEVOTE.